Original research |
Peer reviewed |
Concurrent use of reverse transcription-polymerase chain reaction testing of oropharyngeal scrapings and paired serological testing for detection of porcine reproductive and respiratory syndrome virus infection in sows
Steven B. Kleiboeker, DVM, PhD; James R. Lehman, DVM; Thomas J. Fangman, DVM, MS, Diplomate ABVP-SHM
SBK: Department of Veterinary Pathobiology and the Veterinary Medical Diagnostic Laboratory, University of Missouri, College of Veterinary Medicine, Columbia, MO 65211; JRL: Mid Missouri Animal Consultants, Columbia, MO 65203; TJF: Department of Veterinary Medicine and Surgery, University of Missouri, College of Veterinary Medicine, Columbia, MO 65211; Corresponding author: Steven B. Kleiboeker, Department of Veterinary Pathobiology and the Veterinary Medical Diagnostic Laboratory, University of Missouri, College of Veterinary Medicine, 1600 E Rollins, Columbia, MO 65211; Tel: 573-882-6811; Fax: 573-882-1411; E-mail: KleiboekerS@Missouri.edu.
Kleiboeker SB, Lehman JR, Fangman TJ. Concurrent use of reverse transcription-polymerase chain reaction testing of oropharyngeal scrapings and paired serological testing for detection of porcine reproductive and respiratory syndrome virus infection in sows. J Swine Health Prod. 2002;10(6):251-258. Also available as a PDF.
Summary
Objective: To investigate the use of porcine reproductive and respiratory syndrome virus (PRRSV) reverse transcription-polymerase chain reaction (RT-PCR) on oropharyngeal scrapings concurrently with paired serological testing for detection of PRRSV infection in sows in commercial herds.
Methods: Oropharyngeal scrapings were collected from 191 sows in a 1000-sow, commercial farrow-to-finish herd (Herd A) and from 56 sows in a 900-sow, commercial farrow-to-wean herd (Herd B). Sera were collected from all Herd A sows and 20 Herd B sows. An RT-PCR assay was used to amplify RNA extracted from oropharyngeal scrapings, and a commercial serum ELISA was used to assess PRRSV antibody levels.
Results: Oropharyngeal scrapings from 28.3% of Herd A sows and 19.6% of Herd B sows were RT-PCR-positive for PRRSV. Administration of a killed swine influenza vaccine to 80% of Herd A sows 2 weeks before collection of oropharyngeal scrapings did not influence the rate of PRRSV detection. Sera from the 191 Herd A sows and 20 Herd B sows were negative for PRRSV by virus isolation. Virus isolation detected PRRSV in 36.4% of the RT-PCR-positive sows in Herd B. With RT-PCR results as an indicator of the true PRRSV status of the sow, paired ELISA testing had a sensitivity of 70.4% and a specificity of 49.6%.
Implications: Oropharyngeal scrapings were RT-PCR-positive for PRRSV RNA in aviremic, clinically normal sows and in some sows with PRRSV ELISA sample: positive ratios <0.4. The diagnostic parameters of paired serological testing will likely preclude the use of this method for detecting PRRSV RT-PCR-positive sows.
Keywords: swine, porcine reproductive and respiratory syndrome virus, serology, reverse transcription-polymerase chain reaction
Search the AASV web site for pages with similar keywords.
Received: November 12, 2001
Accepted: February 11, 2002
Porcine reproductive and respiratory syndrome (PRRS) is an important disease of swine that is characterized by respiratory and reproductive disease as well as significant production losses. The etiologic agent is PRRS virus (PRRSV),1 one of the main components of the economically important porcine respiratory disease complex. Currently, there are no uniformly satisfactory approaches to PRRSV control for many herds. Both killed and modified live vaccines often fail to evoke a fully protective immune response or eliminate shedding, especially in the face of challenge with a different (heterologous) strain of PRRSV.2-5 Furthermore, modified live vaccines have limitations because of the potential for persistence within a herd and uncontrolled spread of vaccine virus.6 For example, Christopher-Hennings et al2 demonstrated that vaccine virus (administered in an "extra-label" manner) was shed in the semen of vaccinated boars up to 39 days post-vaccination. As an alternative to vaccination, a "test-and-removal" strategy has been used with success in some herds.7,8 In this approach, serum samples are tested by enzyme-linked immunosorbent assay (ELISA), indirect immunofluorescent assay (IFA), and reverse transcription-polymerase chain reaction (RT-PCR). Sows with a positive result from any one of the three tests are considered to be either acutely or persistently infected and therefore moved off the farm. However, "test and removal" is advocated only in herds that are predominantly seronegative, have no history of recent clinical disease, and have never been vaccinated with a modified live PRRSV vaccine. These herds have no existing PRRSV-induced disease nor current production losses attributable to PRRSV and thus are not representative of many PRRSV-infected swine herds. The final strategy that has been commonly implemented is depopulation followed by repopulation with PRRSV-negative stock. However, due to the required interruption in production, this is not an economically viable option for many producers. Given the limitations of current control strategies and the continued negative economic impact of PRRSV, it is clear that many herds might benefit from additional diagnostic and management approaches that would diminish the effects of this pathogen on swine production.
Persistence of PRRSV in individual pigs and in swine herds is one of the major impediments to PRRS control. After the acute phase of PRRSV infection, which is typically characterized by viremia and clinical disease, many pigs fully recover, yet carry a low-level viral infection for an extended period of time. These "carrier" pigs are persistently infected with PRRSV, shedding the virus either intermittently or continuously, and may infect naive pigs with direct or indirect contact. Under experimental conditions, persistent infection with PRRSV has been well documented.9-14 Most notably, infectious virus has been recovered for up to 157 days post-infection.15 Throughout these studies, efforts have been made to determine the optimal tissue or anatomical site and the optimal technique for identification of persistently infected pigs. Under experimental conditions, an oropharyngeal scraping has been shown to be the superior antemortem sample for detecting persistent infection when compared to serum, tonsillar biopsies, conjunctival swabs, and a number of other tissue samples.15,16 When detection methods have been compared, RT-PCR (insome experiments a nested amplifica-tion strategy was used) detected PRRSV in more samples and at later times post-infection than virus isolation (VI).16,17 However, the value of VI should not be overlooked, as it provides definitive proof that infectious virus was present at the time of sampling. An additional advantage of RT-PCR over both VI and swine bioassay is that the results of RT-PCR may be available within 1 to 3 days after sample collection, as opposed to 1 to 4 weeks for other techniques with similar sensitivity. Thus, experimental evidence indicates that RT-PCR of an oropharyngeal scraping may be the optimal method to rapidly identify persistently infected pigs on an antemortem basis. Unfortunately, the diagnostic sensitivity of this, or any technique, has not been well documented for persistently infected pigs, most often due to a limited number of animals in the studies. In the only study to directly address this question to date, Horter et al16 demonstrated that RT-PCR of an antemortem oropharyngeal scraping could detect a mean of 81% of all persistently infected pigs (as determined by a swine bioassay using a variety of post-mortem samples) for up to 105 days post-inoculation.
In this study, the central hypothesis was that RT-PCR of oropharyngeal scrapings or paired serological testing could be used to identify sows that were potentially persistently infected with PRRSV. Thus, our research objectives were twofold. Our first objective was to investigate the ability of an RT-PCR assay to detect PRRSV RNA in oropharyngeal scrapings collected from sows under typical production conditions. It was reasoned that aviremic sows with RT-PCR-positive oropharyngeal scrapings may be persistently (rather than acutely) infected and thus represent a potential reservoir for PRRSV in the herd. The second objective was to determine if paired serological testing could be used to reliably identify pigs with RT-PCR-positive oropharyngeal scrapings. If a correlation could be made between the results of RT-PCR testing on an oropharyngeal scraping and results of paired serological tests, the latter would be a more useful approach. Practitioners are accustomed to collecting sera and this sample can be submitted for routine analysis to a large number of veterinary diagnostic laboratories.
Materials and methods
Study herds
Samples were collected from sows in two commercial swine herds. The first herd (Herd A) was a 1000-sow, single-site, farrow-to-finish herd. The second herd (Herd B) was a 900-sow, single-site, farrow-to-wean herd. All sows tested for this study were in the breeding-gestation phase of production and were housed either in crates or pens. Both herds had a history of PRRS clinical disease, either in nursery pigs (Herd A) or in the gestational phase of production (Herd B). Modified live PRRS vaccine had last been used in Herd A 6 months prior to the initiation of this study. Herd B had no history of PRRS vaccine use. On Day 14 (after collection of the first serum sample on Day 0), approximately 80% of the sows in Herd A were immunized with a killed H1N1 swine influenza A virus (SIV) vaccine (MaxiFlu; Schering-Plough Animal Health, Union, New Jersey).
Study design
Herd A. Paired serum samples were taken on Day 0 and Day 28 from 191 sows. Oropharyngeal scrapings were collected from the same 191 sows on Day 28. On Day 72, oropharyngeal scrapings were collected from a subset of 28 sows from which the first oropharyngeal scrapings (Day 28) had been negative for PRRSV by RT-PCR. The RNA was extracted from stored (-80°C) serum samples of sows with RT-PCR-positive oropharyngeal scrapings and from 20 randomly selected samples.
Herd B. Single serum samples were collected from 20 sows, and on the same day, oropharyngeal scrapings were collected from 56 sows (including the 20 sows from which serum samples were collected). The RNA was extracted from stored (-80°C) serum samples of all 20 sows and tested by RT-PCR for PRRSV.
Serology
Serum samples were analyzed by the HerdChek PRRS ELISA (IDEXX Laboratories, Westbrook, Maine) according to the manufacturer's instructions, at the University of Missouri's Veterinary Medical Diagnostic Laboratory (Columbia). A sample: positive (S:P) ratio >=0.4 is considered positive for this assay.
Oropharyngeal scrapings
Oropharyngeal samples were collected by aggressively scraping a minimum of three times directly over the entire palatine tonsil with a sterile standard long-handled teaspoon (Utica Cutlery Co, Utica, New York; total length 20.4 cm), held with small locking pliers to provide a firmer grip. Prior to use, all spoons were rinsed with distilled water and autoclaved at 121°C, 15 psi, for 30 minutes. Prior to subsequent use, each spoon was individually washed first in tap water, then in 70% ethyl alcohol, and finally in two rinses with distilled water. Spoons were re-autoclaved after use. To allow optimal sample collection, the oropharynx was visualized by use of an oral speculum and flashlight, and the sows were restrained with a snout snare. The samples collected were considered optimal if cellular debris (presumably from the tonsillar crypts), a slight blood tinge, or both, were visually observed in the sample at the time of scraping. The sample was removed from the spoon with a sterile Dacron swab and placed into 0.5 mL of 10 mM tris [hy-droxymethyl] aminomethand hydrochloride (Sigma Chemicals, St Louis, Missouri), 1 mM disodium ethylenediamine- tetraacetic acid (Sigma Chemicals, St. Louis, Missouri), pH 8.0. The samples were held on ice packs for transportation to the laboratory.
RNA extraction
The oropharyngeal scraping sample collected from each sow in Herd A was individually extracted for RNA using Trizol LS (Life Technologies, Grand Island, New York) according to the manufacturer's instructions. Approximately one-half (0.25 mL) of the oropharyngeal scraping collected from each Herd B sow and 0.25 mL of each serum sample collected from some sows in both herds was used for RNA extraction using the same method. For both serum samples and oropharyngeal scraping samples, 5 mg of tRNA (Gibco BRL/Life Technologies, Grand Island, New York) was added to each sample prior to extraction to facilitate precipitation of RNA. The RNA pellet was resuspended in 6 mL of RNAse-free water. Two mL of each RNA sample was individually amplified by RT-PCR for PRRSV.
RT-PCR amplification
Amplification was performed with the Qiagen One-step RT-PCR kit (Qiagen, Inc, Valencia, California) in a single tube for each sample, with 1.0 mL One-step RT-PCR enzyme mix in the manufacturer's buffer containing 2.5 mM MgCl2 and 0.2 mM (each) deoxynucloetide triphosphates in a final reaction volume of 25 mL, with thermocycling performed in a Perkin-Elmer 9700. Thermocycling conditions were as follows: 50°C (40 minutes), 95°C (12 minutes), followed by 12 cycles of denaturation (95°C, 30 seconds), annealing (72°C, 30 seconds), and extension (72°C, 90 seconds), with the annealing temperature in these cycles reduced by 1°C each cycle. An additional 38 cycles of denaturation (95°C, 30 seconds), annealing (60°C, 30 seconds), and extension (72°C, 90 seconds) were performed, followed by a final extension at 72°C for 7 minutes. Primers used for amplification were (forward) 5´-AGCTGAATGGCACAGATTGG-3´ (reverse) 5´-TGTGGAGCCGTGCTATCAT-3´. These primers correspond to base pair numbers 13914 to 13933 (for the forward primer) and 14384 to 14402 (for the reverse primer) of the prototype US PRRSV isolate VR-2332 (GenBank accession number U87392) and amplified a 489-bp fragment from the open reading frame 5-6 region of the PRRSV genome. Each primer was used at a final concentration of 1.0 mM. These primers were selected to amplify the broadest range of PRRSV isolates feasible on the basis of analysis of hundreds of sequences available in public domain databases. Following amplification, 10 mL of the amplification product was analyzed by agarose gel electrophoresis with visualization by ethidium bromide staining. Weak positive reactions were confirmed by a second round amplification using the same protocol. At each step of sample collection, RNA purification, RT-PCR amplification, and analysis, strict protocols were followed to prevent cross-contamination between samples.
The analytical sensitivity of the PRRSV RT-PCR assay used in this study was determined by addition of dilutions of PRRSV stocks18 titered on MARC-145 cells by plaque assay methodology19 to oropharyngeal scrapings collected from sows in a herd known to be PRRSV-negative. The analytical sensitivity of this assay was not reduced by the oropharyngeal-scraping sample when compared to media controls. The specificity of the RT-PCR assay was evaluated by assaying RNA prepared from oropharyngeal scrapings of sows from a known PRRSV-negative herd. All samples tested were RT-PCR-negative. In addition, the PRRSV RT-PCR assay did not produce a positive result when the template was RNA or DNA purified from characterized stocks of swine influenza A virus (H1N1 and H3N2 strains), porcine circovirus type 1, porcine circovirus type 2, porcine respiratory coronavirus, transmissible gastroenteritis virus, vesicular stomatitis virus (Indiana and New Jersey strains), porcine parvovirus, porcine enterovirus, porcine encephalomyocarditis virus, Mycoplasma spp, and Mycoplasma hyopneumoniae.
Virus isolation
Virus isolation was attempted on 191 serum samples collected on Day 28 from Herd A. Two wells of MARC-145 cells in a 24-well plate (0.2 cm2 per well) were inoculated with 0.25 mL of serum per well for each sample. After a 1- to 2-hour incubation at 37°C in a humidified 5% CO2 incubator, the inoculum was removed, the cell monolayer was rinsed with growth medium, and 0.5 mL of growth medium was added to each well. Growth medium was Dulbecco's modified eagle medium supplemented with 10% fetal bovine serum, 0.1% gentamicin, and 1% amphotericin B (all reagents purchased from Gibco BRL/Life Technologies, Grand Island, New York). The inoculated MARC-145 cells were held at 37C in a humidified 5% CO2 incubator and observed daily for 7 days. Samples were then lysed by a single freeze-thaw cycle, and 20% of the lysed-cell suspension in each well was replated onto MARC-145 cells in a single well of a 24-well plate and observed for 7 days. After 7 days, the freeze-thaw and replating procedure was performed a second time for a total of three passages on MARC-145 cells. Virus isolation procedures were attempted a second time (using samples that had been stored at -80°C) on serum collected from Herd A sows that had RT-PCR-positive oropharyngeal scrapings and from a subset of randomly selected Herd A sows that had RT-PCR-negative oropharyngeal scrapings. In the second analysis of these samples, the cells were stained with the fluoroscein-conjugated anti-PRRSV monoclonal antibody SDOW17 (Rural Technologies, Inc, Brookings, South Dakota) for PRRSV antigen, using standard procedures,20 after the second and third passages on MARC-145 cells. Virus isolation was attempted on serum collected from 20 Herd B sows, and the cells were stained for PRRSV antigen after the second and third passage on MARC-145 cells as described above.
Virus isolation was attempted on approximately one-half of the volume (0.25 mL) from each of the oropharyngeal-scraping samples that were collected from the 56 sows in Herd B. These samples were first freeze-thawed (one cycle of -80°C), then centrifuged at 600g for 30 minutes (4°C). The supernatant was filtered through a 0.45-µm syringe filter, then, because of the small sample volume of the oropharyngeal scraping, an additional 0.5 mL of cell culture media was passed through the filter. Two wells of MARC-145 cells were inoculated for each sample. After a 1- to 2-hour incubation at 37°C in a humidified 5% CO2 incubator, the inoculum was removed, the cell monolayer was rinsed with growth medium, and 0.5 mL of growth medium was added to each well. The inoculated MARC-145 cells were held at 37°C in a humidified 5% CO2 incubator and observed daily for 7 days. Samples were then lysed by a single freeze-thaw cycle, and 20% of the lysed-cell suspension in each well was replated onto a single well of a 24-well plate and observed for 7 days. This procedure was repeated for one additional passage. All inoculated cells were tested for PRRSV antigen by fluorescent antibody staining as described above.
Calculations and statistical analysis
The result of RT-PCR on the oropharyngeal scraping was used as an indication of the PRRSV status of a sow. To calculate the diagnostic parameters of the PRRS ELISA, the paired serological testing of a sow was considered positive if the change in S:P ratio was >0 and negative if the change was <=0.
Statistical analyses were performed by one-way analysis of variance (Statistical Analysis Systems, Release 8.0, Cary, North Carolina) using the general linear model procedure. Mean values for the ELISA S:P ratios were generated by the least squares means method. Differences in the mean ELISA S:P values for sows that were RT-PCR-positive and RT-PCR-negative on oropharyngeal scrapings were determined using Fisher's least significant difference method. Values ofP<=.05 were considered significantly different.
When a portion of samples were selected for additional or secondary testing, the samples were selected through the use of a table of random numbers generated at http://random.org.
Results
RT-PCR assay
For the one-tube RT-PCR assay used in this study, less than 1 plaque forming unit (PFU) could be routinely detected. For further validation, the analytical sensitivity of the one-tube RT-PCR assay described herein was directly compared to the nested RT-PCR assay of Christopher-Hennings et al21 using RNA purified from serial dilutions of quantified PRRSV viral stocks. Both the nested RT-PCR assay and the one-tube assay were consistently capable of detecting less than a single PFU, and neither assay demonstrated a consistent advantage in terms of analytical sensitivity.
RT-PCR of oropharyngeal scrapings and serum
Oropharyngeal scrapings collected from 54 of 191 sows (28.3%) in Herd A on Day 28 were RT-PCR-positive for PRRSV. Of the 54 positive samples, ten were subjectively characterized as strong positive. The remaining positive results were moderate to weak positives, suggesting that for some samples the assay was near the limits of detection. A second round of amplification confirmed all weak positive results, but did not detect any additional positive samples from 22 randomly selected negative reactions.
Of the 191 oropharyngeal scrapings collected in Herd A, 154 were from sows that had been recently immunized with a killed SIV vaccine. For the SIV-vaccinated sows, 43 samples (27.9%) were RT-PCR-positive for PRRSV. For the 37 sows that had not been vaccinated, oropharyngeal scrapings from 11 sows (29.7%) were RT-PCR- positive.
Oropharyngeal scrapings collected on Day 72 from 28 sows that were previously RT-PCR-negative were all RT-PCR-negative for PRRSV. No indication of previous tissue damage was observed at the time that the second oropharyngeal scraping was collected.
Oropharyngeal scrapings collected from 11 of 56 sows (19.6%) in Herd B were RT-PCR-positive for PRRSV.
Extraction of RNA was performed on stored serum from the 54 Herd A sows with RT-PCR-positive oropharyngeal scrapings on Day 28, on 20 randomly selected serum samples from Herd A, and on the 20 serum samples from Herd B. All samples from both herds tested negative for PRRSV by RT-PCR.
Virus isolation
After a total of three passages on MARC-145 cells (ie, initial inoculation and two blind passages), PRRSV was not detected in sera from any of the 191 sows in Herd A or the 20 sows in Herd B. Virus isolation was attempted a second time on a subset of samples, with the additional step of staining for PRRSV antigen using the monoclonal antibody SDOW17 after three passages on MARC-145 cells. Again, PRRSV was not detected in any of the serum samples from Herd A or B.
Cytopathic effect was observed after two or three passages on MARC-145 cells in four of the 56 oropharyngeal scraping samples (7.1%) collected in Herd B. Staining with SDOW17 identified PRRSV antigen in cells inoculated with these four samples. No PRRSV antigen was detected in cells inoculated with the 52 other samples tested from Herd B. Each of the four VI-positive samples was also RT-PCR-positive.
Serology
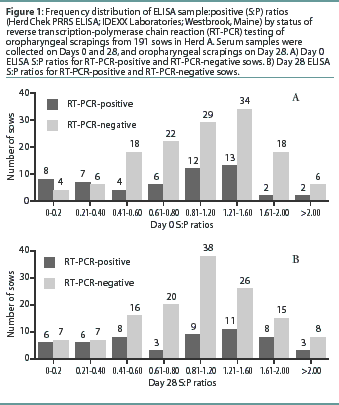
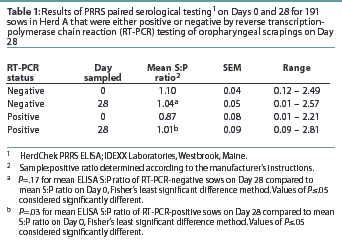
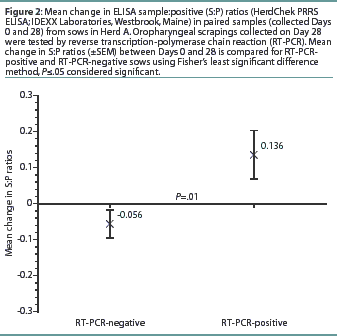
Herd A. The frequency distribution of PRRSV ELISA results for Day 0 and Day 28 serum samples are shown in Figure 1 as a function of status by results of RT-PCR on oropharyngeal scrapings. The means, SEM, and range for the PRRSV ELISA results from Herd A are reported in Table 1. Seventeen of 54 serum samples (31.5%) collected from Herd A sows with RT-PCR-positive oropharyngeal scrapings were negative by PRRSV ELISA on either Day 0 or 28, and 9 of 54 (16.7%) were negative on both Days 0 and 28. Furthermore, 4 of 54 serum samples (7.4%) from the sows with RT-PCR-positive oropharyngeal scrapings had S:P ratios < 0.1 on both Days 0 and 28. When the serological ELISA results from Day 0 and Day 28 serum samples were compared, no difference was observed (P=.17) in the mean S:P ratio in the serum samples collected from sows that had RT-PCR-negative oropharyngeal scrapings (Table 1). In contrast, serum collected from sows that had RT-PCR-positive oropharyngeal scrapings demonstrated a slight increase (P=.03) in mean S:P ratio between Day 0 and Day 28. When the mean change in S:P ratio from Day 0 to Day 28 was compared (Figure 2), the RT-PCR-positive and RT-PCR-negative sows were different (P=.01). Linear regression analysis comparing the results of the RT-PCR on oropharyngeal scrapings to the paired ELISA results demonstrated a correlation between these two assays (r = 0.836).
Herd B. The mean PRRSV ELISA S:P ratio for the 11 sows with RT-PCR-positive oropharyngeal scrapings was 0.59 (SEM = 0.16, range = 0.02 - 1.21), and two of the RT-PCR-positive sows (18.1%) were sero-negative. The mean S:P ratio for the 45 RT-PCR-negative sows was 0.54 (SEM = 0.11, range = 0.01 - 1.28).
The number of RT-PCR-positive and RT-PCR-negative sows with a positive or negative change in S:P ratio is shown in Table 2. The diagnostic characteristics of paired serological testing by PRRS ELISA are shown in Table 3. When the serological results of sows vaccinated against SIV were compared to those not vaccinated for SIV, no difference (P=0.21) was detected (data not shown).


Discussion
Despite more than a decade of research, PRRS remains one of the most economically important diseases of swine.22 In view of the limitations of current control strategies for some herds, new methods and approaches will be required to reduce the economic impact of PRRSV. Elimination of this pathogen from swine herds may be the optimal long-term strategy for PRRSV control. However, the presence of persistently infected "carriers" in a herd may complicate eradication and control efforts, since these pigs can potentially shed the virus months after the acute phase of disease and thus infect naive pigs. The ability to reliably detect persistently infected pigs remains a significant challenge to producers, veterinarians, and researchers in this field.
In this study, we evaluated the ability of a one-tube, non-nested RT-PCR assay performed on RNA extracted from oropharyngeal scrapings of sows to detect PRRSV under typical production conditions. Under controlled experimental conditions, previous work has demonstrated that this was the optimal antemortem sample and method for identification of persistently infected pigs.15,16 This assay was capable of detecting trace amounts (ie, less than 1 PFU) of viral RNA in clinical samples. The ability to detect less than a single infectious viral particle is presumably due to the presence of defective interfering viral particles, which are commonly produced by viruses. Extensive efforts were taken to optimize this assay (through oligonucleotide primer selection and optimization of reagent concentrations and reaction conditions) so that very small amounts of PRRSV RNA could be detected from clinical samples without the need for nested amplification. Additionally, the nucleotide sequences of numerous PRRSV strains (available in the public domain database) were analyzed for primer selection so that the broadest range possible of isolates could be detected without compromising diagnostic specificity. While numerous reports have detailed the use of RT-PCR for diagnosis of PRRSV,21, 23-31 many of these assays would fail to detect a broad range of PRRSV isolates (on the basis of analysis of public-domain databases) or would depend on nested amplification, Southern hybridization, or both for reliable detection of very small amounts of PRRSV (ie, less than a single PFU). In addition to the potential risk of laboratory-related false-positive results due to contamination associated with a nested-amplification strategy, both a second round of amplification and Southern hybridization increase the time from sample submission to reporting of test results and increase the expense of sample analysis.
In this study, a high proportion of oropharyngeal scrapings from sows were RT-PCR-positive for PRRSV, although none of these sows were viremic by VI or RT-PCR at the time of oropharyngeal sampling. Four of the 56 oropharyngeal scraping samples collected in Herd B were positive for PRRSV by VI, and each of these four samples was also positive for PRRSV RNA by RT-PCR. For the purpose of the present study, it was reasoned that clinically normal, aviremic sows in which the oropharyngeal scraping was RT-PCR-positive had the potential to be persistently infected with PRRSV. In view of the level of clinical disease attributable to PRRSV in both herds, the proportion of RT-PCR-positive sows was not unexpected. However, a significant limitation of this study is that additional testing (eg, transmission experiments) were not performed to confirm that PRRSV could be transmitted from the RT-PCR-positive sows, and that the RT-PCR-negative sows were free of infectious virus.
Although collection of the oropharyngeal scraping was judged to be relatively quick (2 to 3 minutes per sow) and straightforward in this study, collection of each sample required the efforts of three people, similar to collection of a tonsillar biopsy for PRRSV detection.32 In contrast to a tonsillar biopsy, however, the oropharyngeal scraping caused no apparent trauma or tissue damage to the sow, even when cellular debris or a blood-tinge was noted in the scraped material. Additionally,the method of collection of the oropharyngeal scraping allowed direct and clear visualization of the palatine tonsil. Thus, sample collection was judged to be accurate, with each sample containing ample amounts of cellular material, presumably expressed directly from the tonsillar crypts.
As a second objective, we compared paired PRRSV ELISA test results to the results of RT-PCR testing of the oropharyngeal scrapings. Comparison of the proportion of RT-PCR-positive sows to the proportions with positive and negative changes in S:P ratio suggested a slight correlation between these two assays. However, it is our conclusion that the diagnostic parameters of paired serological testing severely limited its usefulness in consistently identifying sows that were RT-PCR-positive and thus potentially persistently infected. While the sensitivity and negative predictive value of the paired ELISA results relative to RT-PCR of an oropharyngeal scraping were reasonably high, the specificity and positive predictive value were very low. Thus, on the basis of results of paired serological testing, only half of the true-negative sows would be correctly identified, and two-thirds of the sows with a rising S:P ratio would be incorrectly identified as being potentially persistently infected with PRRSV. One additional observation made from the serology results of this study was that S:P values from a single sample provided little information concerning the PRRSV status of sows that might have been persistently infected with PRRSV. For example, a portion of serum samples collected from sows with RT-PCR-positive oropharyngeal scrapings in both Herds A and B had ELISA S:P ratios <0.4. Thus, ELISA results in either vaccinated or unvaccinated herds may not consistently provide useful information concerning the current PRRSV RT-PCR status of an individual sow.
In this study, we used a PRRSV RT-PCR assay capable of detecting low concentrations of PRRSV RNA in oropharyngeal scrapings of clinically normal, aviremic sows from PRRSV-positive herds under field conditions. Future studies will utilize necropsy with extensive post-mortem tissue sampling and transmission trials to assess the diagnostic accuracy of this RT-PCR assay on oropharyngeal scrapings, with respect to its ability to identify persistently infected sows that may shed PRRSV under production conditions. This assay could provide the foundation for an on-farm PRRSV eradication strategy. At the very least, an antemortem assay able to detect sows persistently infected with PRRSV will allow a more direct assessment of the role these animals play in PRRSV spread within a herd.
Implications
- PRRSV was detected by RT-PCR in a high proportion of oropharyngeal scrapings collected from aviremic sows in endemically-infected herds.
- RT-PCR of oropharyngeal scrapings allowed rapid and sensitive antemortem identification of sows that may be persistently infected with PRRSV.
- The results of paired PRRS HerdChek ELISA tests did not correlate strongly with the PRRSV RT-PCR results.
- A portion of sows with RT-PCR-positive oropharyngeal scrapings had PRRS ELISA S:P ratios <0.4.
- SIV vaccination did not increase the incidence of PRRSV RNA detection in oropharyngeal scrapings of sows.
Acknowledgements
The authors wish to thank Drs Jennifer Keaton, Monica Otto, Elizabeth Rhinehart, and Daniel Thebold for invaluable assistance in sample collection. Dr Mark Ellersieck performed statistical analysis of the serological results. Finally, the authors are extremely grateful to the swine producers who allowed us access to their herds for sample collection.
References - refereed
1. Plagemann PGW. Lactate dehydrogenase-elevating virus and related viruses. In: Fields B, Knipe DM, Howley PM, eds. Fields Virology, 3rd ed. Philadelphia, Pennsylvania: Lippincott Williams & Wilkins; 1996:1105-1120.
2. Christopher-Hennings J, Nelson EA, Nelson JK, Benfield DA. Effects of a modified-live virus vaccine against porcine reproductive and respiratory syndrome in boars. Am J Vet Res. 1997;58:40-45.
3. Lager KM, Mengeling WL, Brockmeier SL. Evaluation of protective immunity in gilts inocu-lated with the NADC-8 isolate of porcine reproductive and respiratory syndrome virus (PRRSV) and challenge-exposed with an antigenically distinct PRRSV isolate. Am J Vet Res. 1999;60:1022-1027.
4. Mengeling WL, Lager KM, Vorwald AC. Safety and efficacy of vaccination of pregnant gilts against porcine reproductive and respiratory syndrome. Am J Vet Res. 1999;60:796-801.
5. Meng XJ. Heterogeneity of porcine reproductive and respiratory syndrome virus: implication for current vaccine efficacy and future vaccine development. Vet Micro. 2000;74:309-329.
6. Mengeling WL, Vorwald AC, Lager KM, Clouser DF, Wesley RD. Identification and clinical assessment of suspected vaccine-related field strains of porcine reproductive and respiratory syndrome virus. Am J Vet Res. 1999;60:334-340.
7. Dee SA, Moliter TW. Elimination of PRRS virus using test and removal process. Vet Rec. 1998; 143:474-476.
8. Dee SA, Moliter TW, Rossow KD. Epidemiological and diagnostic observations following the elimination of PRRS virus from a breeding herd of pigs by the test and removal protocol. Vet Rec. 2000;146:211-213.
9. Albina E, Madec F, Cariolet R, Torrison J. Immune response and persistence of the porcine reproductive and respiratory syndrome virus in infected pigs and farm units. Vet Rec. 1994;134:567-573.
10. Allende R, Laegreid WW, Kutish GF, Galeota JA, Wills RW, Osorio FA. Porcine reproductive and respiratory syndrome virus: Description of persistence in individual pigs upon experimental infection. J Virol. 2000;74:10834-10837.
12. Christopher-Hennings J, Nelson EA, Hines RJ, Nelson JK, Swenson SL, Zimmerman JJ, Chase CCL, Yaeger MJ, Benfield DA. Persistence of porcine reproductive and respiratory syndrome virus in serum and semen of adult boars. J Vet Diagn Invest. 1995;7:456-464.
13. Sur JH, Cooper VL, Galeota JA, Hesse RA, Doster AR, Osorio FA. In vivo detection of porcine reproductive and respiratory syndrome virus RNA by in situ hybridization at different times postinfection. J Clin Micro 1996;34:2280-2286.
15. Wills RW, Zimmerman JJ, Yoon KJ, Swenson SL, McGinely MJ, Hill HT, Platt KB, Chistopher-Hennings J, Nelson EA. PRRS virus: a persistent infection. Vet Micro. 1997;55:231-240.
16. Horter DC, Pogranichiy RM, Chang CC, Evans RB, Yoon KJ, Zimmerman JJ. Characterization of the carrier state in porcine reproductive and respiratory syndrome virus infection. Vet Micro. 2002;86:213-228.
18. Unthum AR, Mengeling WL. Restriction fragment length polymorphism analysis of strains of porcine reproductive and respiratory syndrome virus by use of a nested-set reverse transcription-polymerase reaction. Am J Vet Res. 1999;60:802-806.
19. Burleson FG, Chambers TM, Wiedbrauk DL. Virology: a laboratory manual. San Diego, California: Academic Press; 1992:74-84.
20. Hsiung GD, Fong CKY, Landry ML. Diagnostic Virology, 4th ed. New Haven, Connecticut: Yale University Press; 1994:56-58.
21. Christopher-Hennings J, Nelson EA, Nelson JK, Hines RJ, Swenson SL, Hill HT, Zimmerman JJ, Katz JB, Yaeger MJ, Chase CC, Benfield DA. Detection of porcine reproductive and respiratory syndrome virus in boar semen by PCR. J Clin Micro. 1995;33:1730-1734.
23. Cheon DS, Chae C. Comparison of virus isolation, reverse transcription-polymerase chain reaction, immunohistochemistry, and in situ hybridization for the detection of porcine reproductive and respiratory syndrome virus from naturally aborted fetuses and stillborn piglets. J Vet Diagn Invest. 2000;12:582-587.
24. Guarino H, Goyal SM, Murtaugh MP, Morrison RB, Kapur V. Detection of porcine reproductive and respiratory syndrome virus by reverse transcription-polymerase chain reaction using different regions of the viral genome. J Vet Diagn Invest. 1999;11:27-33.
25. Larochelle R, Magar R. Evaluation of the presence of porcine reproductive and respiratory syndrome virus in packaged pig meat using virus isolation and polymerase chain reaction (PCR) method. Vet Micro. 1997;58:1-8.
26. Mardassi H, Wilson L, Mounir S, Dea S. Detection of porcine reproductive and respiratory syndrome virus and efficient differentiation between Canadian and European strains by reverse transcription and PCR amplification. J Clin Micro. 1994;32:2197-2203.
27. Oleksiewicz MB, Botner A, Madsen KG, Storgaard T. Sensitive detection and typing of porcine reproductive and respiratory syndrome virus by RT-PCR amplification of whole viral genes. Vet Micro. 1998;64:7-22.
28. Spagnuolo-Weaver M, Walker IW, McNeilly F, Calvert V, Graham D, Burns K, Adair BM, Allan GM. The reverse transcription polymerase chain reaction for the diagnosis of porcine reproductive and respiratory syndrome: comparison with virus isolation and serology. Vet Micro. 1998;62:207-215.
29. Suarez P, Zardoya R, Prieto C, Solana A, Tabares E, Bautista JM, Castro JM. Direct detection of the porcine reproductive and respiratory syndrome (PRRS) virus by reverse polymerase chain reaction (RT-PCR). Arch Virol. 1994;135:89-99.
30. Van Woensel P, Van der Wouw J, Visser N. Detection of porcine reproductive respiratory syndrome virus by the polymerase chain reaction. J Virol Methods. 1994;47:273-278.
31. Wagstrom EA, Yoon KJ, Cook C, Zimmerman JJ. Diagnostic performance of a reverse transcription-polymerase chain reaction test for porcine reproductive and respiratory syndrome virus. J Vet Diagn Invest. 2000;12:75-78.
References - non refereed
11. Benfield DA, Nelson J, Rossow KD, Rowland RR, Lawson SR, Steffen M, Collins M. Pathogenesis and persistence of PRRS. Proc AD Leman Swine Conf. 1998:169-171.
17. Wills RW, Doster AR, Galeota J, Sur JH, Osorio FA. Infection duration and proportion of persistently PRRS virus infected pigs. Conf Res Workers An Dis. 1999;80:178.
22. Polson DD, Marsh WE, Dial GD, Christianson WT. Financial impact of Porcine Epidemic Abortion and Respiratory Syndrome (PEARS); Proc 12th IPVS Conf. 1992:132.