| Diagnostic notes | Peer reviewed |
Cite as: Hause BM, Duff JW, Scheidt A, et al. Virus detection using metagenomic sequencing of swine nasal and rectal swabs. J Swine Health Prod. 2016;24(6):304–308.
Also available as a PDF.
SummaryAdvances in DNA sequencing have increased our ability to generate large amounts of sequence data at lower costs. These developments have enabled microbial detection and characterization directly from clinical specimens, known as metagenomic sequencing. Viral metagenomic sequencing was performed on five nasal- and five fecal-swab pools collected from each of two primary and two secondary market slaughterhouses and a cull-swine buying station in the southeastern United States. Sequences were assembled de novo and analyzed by BLASTN to identify viruses present in the samples. Twenty seven different viruses were identified. Reads similar to a diverse family of single-stranded circular DNA viruses were identified in nearly every sample (47 of 50). Other viruses identified at all five sampling sites and in over half of the samples were bocavirus, torovirus, posavirus, torque teno virus, IAS virus, picobirnavirus, and teschovirus. Viruses identified in multiple sites in greater than 20% of the samples included enterovirus, parvovirus, influenza A virus, sapelovirus, and Senecavirus A. Other significant swine viruses detected less frequently include porcine circovirus type 2, porcine epidemic diarrhea virus, and porcine deltacoronavirus. Together, these results suggest that metagenomic sequencing is a powerful tool for virus detection and characterization. | ResumenLos avances en la secuenciación de DNA han incrementado nuestra habilidad para generar grandes cantidades de información de secuencias a costos más bajos. Estos desarrollos han permitido la caracterización y la detección microbiana directamente de especímenes clínicos, conocida como secuenciación metagenómica. Se realizó la secuenciación metagenómica viral en cinco grupos de cinco muestras nasales y fecales recolectadas de dos mataderos primarios y dos secundarios y de una estación de compra de animales de desecho en el sureste de los Estados Unidos. Las secuencias se montaron de novo y se analizaron por medio de BLASTN para identificar los virus presentes en las muestras. Se identificaron veintisiete virus diferentes. Se identificaron lecturas semejantes a una familia diversa de virus de DNA circular de cadena simple casi en cada muestra (47 de 50). Otros virus identificados en los cinco sitios de muestreo y en más de la mitad de las muestras fueron bocavirus, torovirus, posavirus, virus torque teno, virus IAS, picobirnavirus, y teschovirus. Los virus identificados en sitios múltiples en más del 20% de las muestras incluyeron enterovirus, parvovirus, virus de la influenza A, sapelovirus, y Senecavirus A. Otros virus porcinos significativos detectados con menor frecuencia incluyeron circovirus porcino tipo 2, virus de la diarrea epidémica porcina, y el deltacoronavirus porcino. Conjuntamente, estos resultados sugieren que la secuenciación metagenómica es una herramienta poderosa para la caracterización y detección de virus. | ResuméLes avancées dans le séquençage de l’ADN ont augmenté la capacité à générer de grandes quantités de données de séquences à des coûts moindres. Ces développements ont permis la détection microbienne et la caractérisation directement à partir de spécimens cliniques, connus sous l’appellation de séquençage métagénomique. Le séquençage métagénomique viral a été effectué sur cinq pools d’écouvillons nasaux et cinq pools d’écouvillons rectaux prélevés de chacun de deux abattoirs primaires et deux abattoirs secondaires, ainsi que d’une station d’achat d’animaux réformés dans le sud-est des États-Unis. Les séquences ont été assemblées de novo et analysées par BLASTN afin d’identifier les virus présents dans les échantillons. Vingt-sept virus différents ont été identifiés. Des lectures similaires à une famille variée de virus à ADN circulaire simple brin ont été identifiées dans presque tous les échantillons (47 des 50). Les autres virus identifiés dans tous les sites échantillonnés et dans plus de la moitié des échantillons étaient des bocavirus, des torovirus, des posavirus, des torque teno virus, les virus IAS, les picobirnavirus, et les teschovirus. Les virus identifiés dans des sites multiples dans plus de 20% des échantillons incluaient les enterovirus, les parvorirus, le virus de l’influenza A, les sapelovirus, et le Senecavirus A. Les autres virus porcins significatifs détectés moins fréquemment incluaient le circovirus porcin de type 2, le virus de la diarrhée épidémique porcine, et le deltacoronavirus porcin. Ces résultats suggèrent que le séquençage métagénomique est un outil puissant pour la détection et la caractérisation des virus. |
Keywords: swine, DNA sequencing, virus, Senecavirus A, virome
Search the AASV web site
for pages with similar keywords.
Received: December 30, 2015
Accepted: April 15, 2016
For greater than a decade, DNA sequencing has been widely used by swine veterinarians to study the epidemiology of clinically important viruses such as porcine reproductive and respiratory syndrome virus (PRRSV) and influenza A virus (IAV). Traditional DNA sequencing methodology, developed by Sanger, has the advantages of low cost, fast turnaround times, and relatively long read lengths.1 Commonly, the gene encoding the most variable immunodominant viral protein is targeted. Polymerase chain reaction (PCR) is used to amplify the gene of interest, and the purified PCR product is subjected to Sanger sequencing using a DNA primer that binds to the PCR product. Multiple sequencing reads are utilized to achieve complete gene coverage. Prices charged by veterinary diagnostic laboratories in the United States vary, but are typically approximately $100 to $180 per sample, with results available within days. A significant disadvantage of Sanger sequencing is a requirement for a priori knowledge of an organism’s presence in a sample, as well as sufficient sequence homology to enable binding of the sequencing primer.
New DNA sequencing technologies have become more commonplace in diagnostic laboratories.2,3 Next-generation sequencing platforms generate massive amounts of sequencing information at low cost. Owing to this feature, metagenomic sequencing techniques have been developed that utilize random priming approaches to non-specifically amplify nucleic acids present in a sample.4 While these techniques lack specificity and amplify host, environmental, and pathogen sequences, the sheer quantity of sequencing reads enables identification and characterization of pathogen nucleic acids present in relatively low concentrations. For viral metagenomic sequencing, a variety of protocols have been published to enrich the sample for viral populations.5,6 A significant advantage of metagenomic sequencing is the lack of requirement for prior knowledge of virome composition for DNA sequencing. Universal metagenomic sequencing protocols have been published that are capable of detecting both single- and double-stranded DNA and RNA viruses.5 Despite these advantages, next-generation sequencing remains more expensive, on a per-sample basis, than traditional methods, and requires more time. Owing to the large number of sequences generated per sample, data management is more complex, often requiring specialized software.
Large numbers of viruses are known to infect swine, and new viruses are routinely being discovered. While several publications have explored the swine virome, we have limited understanding of the clinical significance of most of these viruses.7,8 The goal of this study was to characterize the swine virome at points of animal concentration and commingling.
Sampling protocol
Nasal and fecal swabs were collected in universal viral transport medium from five individual pigs derived from a single producer and assembled into nasal- and fecal-swab pools. Pigs from five producers were collected per site. Five sites in total were sampled in August 2015. Samples were collected by a veterinarian, and all animals were clinically healthy. Sites 1 and 2 were abattoirs that purchased top-quality hogs (primary market). Sites 3 and 4 were cull-swine abattoirs. Site 5 was a cull-swine buying station. Animals from sites 1 to 4 were greater than 20 weeks of age, while animals at Site 5 were greater than 10 weeks of age.
Metagenomic sequencing
Metagenomic sequencing was performed approximately as previously described.5 Swabs in transport medium were vortexed, and pools of five nasal or fecal swabs were assembled (10 total pools per site). Samples were clarified by centrifugation and subsequently enriched for viral nucleic acids by treatment with a mixture of nucleases.6 Viral nucleic acids were extracted using the MinElute Virus Spin Filter Kit (Qiagen, Valencia, California) according to the manufacturer’s instructions. First-strand cDNA was synthesized from viral RNA using the Superscript III First-Strand Synthesis System (ThermoFisher Scientific, Waltham, Massachusetts) as specified by the manufacturer, with previously described random primers.4 Second-strand synthesis was performed with Sequenase 2.0 DNA Polymerase (ThermoFisher Scientific) followed by cDNA purification using the Agencourt AMPure XP beads (Beckman Coulter, Brea, California). The double-stranded cDNA was amplified with TaKaRa DNA polymerase (Clontech Laboratories, Mountain View, California) with primers identical to those used for first-strand synthesis, but lacking the random hexamer. Size selection and purification were performed using Agentcourt AMPure XP beads, selecting for products > 300 bp. Amplicons were quantified using a Qubit fluorometer (ThermoFisher Scientific), and libraries were prepared by the standard Nextera XT Library Preparation Kit (Illumina, San Diego, California) protocol. All 50 libraries were pooled and sequenced using paired 300-bp reads on a single Miseq (Illumina) run. Sequence reads were parsed using barcodes incorporated during library preparation and imported into CLC Genomics Workbench (CLC Bio, Waltham, Massachusetts). Reads were mapped to the host genome (Sus scrofa), and from the unmapped reads, contigs were assembled de novo. Contigs, the consensus sequences derived from overlapping DNA sequences, were analyzed by the basic local alignment search tool nucleotides (BLASTN). Contigs with expectation (E) values (a measure of database hit strength) less than 10-10 were considered positive for virus identification. For viruses with multiple BLASTN hits to similar viral species, virus identity was assigned to a higher, more inclusive taxonomic level, typically the genus.
Results
Viruses detected in primary market swabs are shown in Figure 1. A total of 19 different viruses were detected. Contigs with significant E values to bocavirus, torovirus, posavirus, IAS virus, picobirnavirus, and a diverse family of circular single-stranded DNA viruses (ssDNA) were found in over half of the primary market swabs. Torque teno virus, teschovirus, and adeno-associated virus were also identified at both sites. Other viruses identified at only one of the primary swine markets include porcine circovirus type 2 (PCV2), astrovirus, enterovirus, porcine parvovirus, parecho-like virus, pasivirus, IAV, sapelovirus, calicivirus, and porcine adenovirus 5. Averages of 6.6 and 5.3 viruses were detected per swab pool from sites 1 and 2, respectively.
Figure 1: Percentage of pooled swab samples collected from primary-market abattoirs positive for identified viruses. Separate nasal- and fecal-swab pools were assembled from swabs collected from five individual pigs derived from a single producer. Pigs from five producers were collected per site. Viral metagenomic sequencing was performed on the five nasal-swab and five fecal-swab pools collected at each site to identify viruses present in the pooled samples.
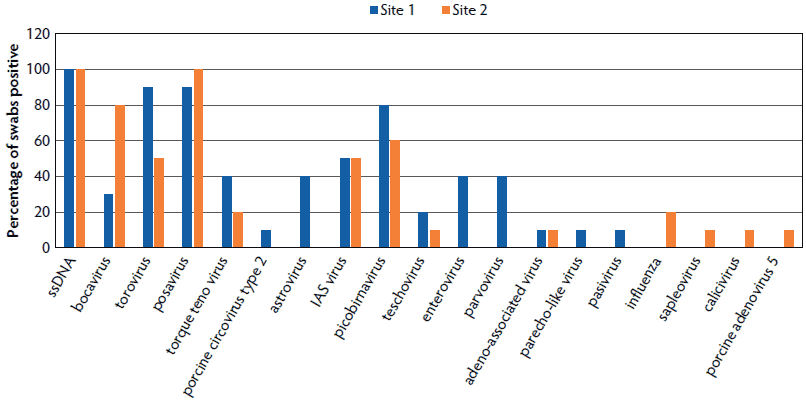
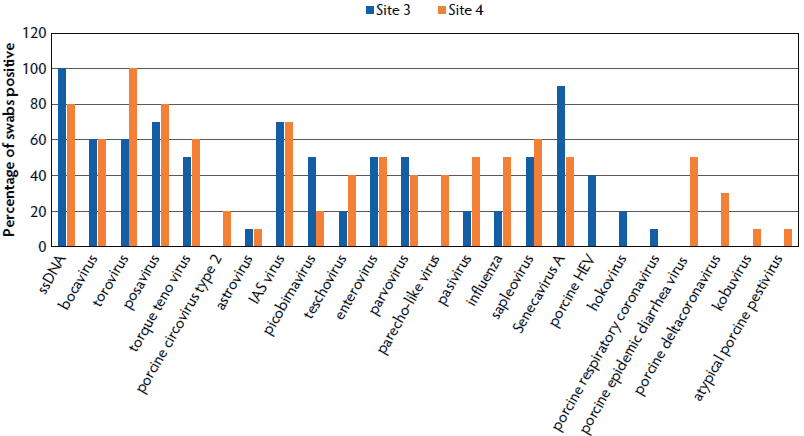
Twenty-four viruses were identified in swabs collected at the two secondary-market abattoirs, nine of which were in over 50% of the swabs (ssDNA, bocavirus, torovirus, posavirus, torque teno virus, IAS virus, enterovirus, sapelovirus, and Senecavirus A; Figure 2). Viruses identified at both secondary sites at lower prevalence include astrovirus, picobirnavirus, teschovirus, parvovirus, pasivirus, and IAV. Viruses identified at a single site were PCV2, parecho-like virus, hemagglutinating encephalomyelitis virus (HEV), hokovirus, porcine respiratory coronavirus, porcine epidemic diarrhea virus (PEDV), porcine deltacoronoavirus, kobuvirus, and atypical porcine pestivirus (APPV). Swab pools from sites 3 and 4 had the highest average number of viruses detected (8.4 and 9.8, respectively).
Figure 2: Percentage of pooled swab samples collected from secondary-market slaughterhouses positive for identified viruses. Study described in Figure 1. Porcine HEV = porcine hemagglutinating encephalomyelitis virus.
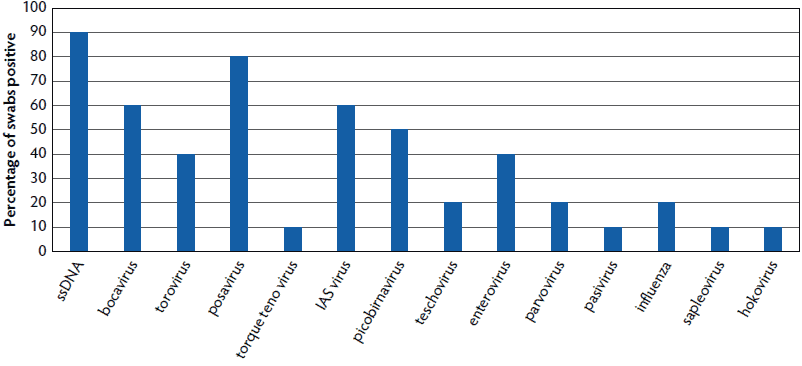
Analysis of swabs from Site 5 identified 14 viruses (Figure 3). The most commonly identified viruses were ssDNA, bocavirus, posavirus, IAS virus, picobirnavirus, enterovirus, and torovirus, similar to those seen at sites 1 to 4. The remaining viruses were also variably identified at sites 1 to 4. An average of 5.2 viruses were identified per swab pool.
Figure 3: Percentage of pooled swab samples collected from a cull-market buying station positive for identified viruses. Study described in Figure 1.
Discussion
In this study, metagenomic sequencing methodology was applied to nasal and fecal swabs collected from swine commingled at slaughter and buying facilities. While the animals were suitable for slaughter, no information was available on the herd-of-origin status or health history. The objective of this study was to identify viruses circulating in swine at animal concentration points. Many of these facilities located in the southeastern United States are in close proximity to swine confinement operations and utilize common transport. Contaminated swine transport was previously implicated as one source of the rapid dissemination of PEDV throughout the United States.9
A minimum of 27 different viruses were identified in nasal and fecal swabs, with an overall average of 7.1 viruses per swab pool. As metagenomic sequencing often yields only partial viral genome sequences, annotation of viruses to a strain or species level can be difficult due to varying homology between different regions of the genomes. Consequently, for some viral genera with multiple species, viruses were identified only to the genus level. For example, six proposed species of porcine parvovirus are known to circulate in the United States.10-12 While BLASTN analysis of contig sequences returned hits to numerous porcine parvovirus species, we could not confidently assign a species. Similarly, some viruses, such as small single-stranded circular DNA viruses, are extremely diverse and often poorly characterized.13
A significant advantage of metagenomic sequencing, compared to conventional detection technologies, is its ability to detect viruses without prerequisite sequence information. This ability uniquely positions this technology for detection of emerging viruses for which methods do not exist or are insufficient. One swab pool was positive for APPV, a highly divergent pestivirus, which was only recently identified and characterized by metagenomic sequencing of swine serum samples.14
As seen in this study, metagenomic sequencing often identifies viruses for which little information is known. For example, IAS virus was identified by metagenomic sequencing of stool samples from humans infected with human immunodeficiency virus (HIV) and with unexplained diarrhea.15 To the knowledge of the authors, this is the first identification of IAS virus in pigs. Likewise, posaviruses have previously been identified in swine feces, but it is unclear if they infect pigs or are merely present in the environment.8 A majority of the remaining viruses are known to infect pigs, with unknown clinical significance. The ability to detect environmental viruses and viruses with unclear etiological roles can complicate interpretation of clinical results, but can serve as a basis for unraveling the complex pathogeneses of disease syndromes. Metagenomic sequencing of diseased and healthy controls in a case-control format has been utilized to identify viruses associated with bovine respiratory disease, followed by quantitative PCR and statistical correlations with clinical signs.16 Alone, metagenomic sequencing will not establish microorganism disease causation; however, it can guide further diagnostic testing to unravel disease etiology. Numerous authors have proposed revisions to Koch’s postulates to establish microorganism causality with disease, taking into account advances in detection technologies.17 Detection of a microorganism in most cases of disease, preferentially in diseased tissues, along with a lack of or lower numbers of microorganisms in healthy controls or unaffected tissues, have been proposed as guidelines for establishing causality of disease with microorganism detection.17 Reduction in the number of microorganisms detected should also correspond with disease resolution.
Senecavirus A (SVA) has caused sporadic outbreaks of vesicular disease in the United States for several decades. Senecavirus A was recently identified in Brazilian pigs with vesicular disease in addition to high neonatal mortality.18,19 Numerous outbreaks of SVA in US pigs were reported in the summer of 2015.20 Besides vesicular disease, high neonatal mortality, resembling clinical signs reported in Brazil, were observed. Polymerase chain reaction assays for SVA performed on 2033 oral-fluid samples from routine diagnostic submissions from 25 states identified five positive cases (1%).21 In this study, SVA was detected in 14 of 20 swab pools (70%) collected from secondary market abattoirs and not detected in primary-market abattoir samples or the buying station. Additional testing is needed to determine if cull animals are reservoirs for SVA. These results also raise the question regarding the association of SVA with animals of a lower health status.
These results demonstrate that metagenomic sequencing is a powerful tool for virus identification and characterization. More widespread use will significantly expand our knowledge of viral epidemiology and likely lead to the discovery of novel agents. Metagenomic sequencing has a number of uses, including identifying viruses in diagnostic samples where traditional diagnostics failed to identify pathogens, determining viral genome sequences directly from clinical samples, profiling viruses present in material used to inoculate other animals, and investigating the viral ecology of disease complexes.
While nasal and fecal swabs were analyzed here for simplicity of collection, choice of samples for sequencing should be based on detection of clinical signs. Metagenomic sequencing is offered as a diagnostic test at the Kansas State Veterinary Diagnostic Laboratory.
Implications
- Under the conditions of this study, viral metagenomic sequencing can identify large numbers of viruses in swine nasal and fecal swabs.
- Metagenomic sequencing can be used to characterize viruses present in clinical samples as part of diagnostic investigations.
Acknowledgements
The authors would like to thank the animal markets for allowing us to conduct our study. Funding for this work was provided in part by Boehringer Ingelheim Vetmedica, Inc, and the Kansas State Veterinary Diagnostic Laboratory.
Conflict of interest
None reported.
Disclaimer
Scientific manuscripts published in the Journal of Swine Health and Production are peer reviewed. However, information on medications, feed, and management techniques may be specific to the research or commercial situation presented in the manuscript. It is the responsibility of the reader to use information responsibly and in accordance with the rules and regulations governing research or the practice of veterinary medicine in their country or region.
References
1. Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA. 1977;74:5463–5467.
2. Blomström AL. Viral metagenomics as an emerging and powerful tool in veterinary medicine. Vet Q. 2011;31:107–114.
3. Quinones-Mateu ME, Avila S, Reyes-Teran G, Martinez MA. Deep sequencing: becoming a critical tool in clinical virology. J Clin Virol. 2014;61:9–19.
4. Allander T, Tammi MT, Eriksson M, Bjerkner A, Tiveljung-Lindell A, Andersson B. Cloning of a human parvovirus by molecular screening of respiratory tract samples. Proc Natl Acad Sci. 2005;102:12891–12896.
5. Hause BM, Collin EA, Anderson J, Hesse RA, Anderson G. Bovine rhinitis viruses are common in U.S. cattle with bovine respiratory disease. PLoS ONE. 2015;10:e0121998.
6. Neill JD, Bayles DO, Ridpath JF. Simultaneous rapid sequencing of multiple RNA virus genomes. J Virol Methods. 2014;201:68–72.
7. Zhang B, Tang C, Yue H, Ren Y, Song Z. Viral metagenomics analysis demonstrates the diversity of viral flora in piglet diarrheic feces in China. J Gen Virol. 2014;95:1603–1611.
8. Shan T, Li L, Simmonds P, Wang C, Moeser A, Delwart E. The fecal virome of pigs on a high-density farm. J Virol. 2011;85:11697–11708.
9. Lowe J, Gauger P, Harmon K, Zhang J, Connor J, Yeske P, Loula T, Levis I, Dufresne L, Main R. Role of transportation in spread of porcine epidemic diarrhea virus infection, United States. Emerg Infect Dis. 2014;20:872–874.
10. Opriessnig T, Xiao CT, Gerber PF, Halbur PG. Identification of recently described porcine parvoviruses in archived North American samples from 1996 and association with porcine circovirus associated disease. Vet Microbiol. 2014;173:9–16.
11. Schirtzinger EE, Suddith AW, Hause BM, Hesse RA. First identification of porcine parvovirus 6 in North America by viral metagenomic sequencing of serum from pigs infected with porcine reproductive and respiratory syndrome virus. Virol J. 2015;12:170. doi: 10.1186/s12985-015-0401-6.
12. Ni J, Qiao C, Han X, Han T, Jang W, Zi Z, Cao Z, Zhai X, Cai X. Identification and genomic characterization of a novel porcine parvovirus (PPV6) in China. Virol J. 2014;11:203. doi: 10.1186/s12985-014-0203-2.
13. Li L, Kapoor A, Slikas B, Bamidele OS, Wang C, Shaukat S, Masroor MA, Wilson ML, Ndjango JN, Peeters M, Gross-Camp ND, Muller MN, Hahn BH, Wolfe ND, Triki H, Bartkus J, Zaidi SZ, Delwart E. Multiple diverse circoviruses infect farm animals and are commonly found in human and chimpanzee feces. J Virol. 2010;84:1674–1682.
14. Hause BM, Collin EA, Peddireddi L, Yuan F, Chen Z, Hesse RA, Gauger PC, Clement T, Fang Y, Anderson G. Discovery of a novel putative atypical porcine pestivirus in pigs in the USA. J Gen Virol. 2015;96:2994–2998.
15. Oude Munnink BB, Canuti M, Deijs M, de Vries M, Jebbink MF, Rebers S, Molenkamp R, van Hemert FJ, Chung K, Cotton M, Snijders F, Sol CJ, van der Hoek L. Unexplained diarrhea in HIV-1 infected individuals. BMC Infect Dis. 2014;14:22. doi: 10.1186/1471-2334-14-22.
16. Ng TF, Kondov NO, Deng X, Van Eenennaam A, Neibergs HL, Delwart E. A metagenomics and case-control study to identify viruses associated with bovine respiratory disease. J Virol. 2015;89:5340–5349.
17. Fredricks DN, Relman DA. Sequence-based identification of microbial pathogens: a reconsideration of Koch’s postulates. Clin Microbiol Rev. 1996;9:18–33.
18. Vannucci FA, Linhares DCL, Barcellos DESN, Lam HC, Collins J, Marthaler D. Identification and complete genome of Seneca Valley virus in vesicular fluid and sera of pigs affected with idiopathic vesicular disease, Brazil. Transbound Emerg Dis. 2015;62:589–593. doi: 10.1111/tbed.12410.
19. Leme RA, Zotti E, Alcântara BK, Oliveira MV, Freitas LA, Alfieri AF, Alfieri AA. Senecavirus A: an emerging vesicular infection in Brazilian pig herds. Transbound Emerg Dis. 2015;62:603–611.
*20. Rademacher C, Madson D, Main R, Yoon KJ, Linhares D, Pineyro P, Canning P, Bates J, Cannon A, Halbur P. AASV News Archive. Senecavirus A (Seneca Valley Virus) in Swine – Breeding Herd Cases. Available at https://www.aasv.org/documents/SVVinSowFarms9215.pdf. Accessed 1 July 2016.
*21. Swine Health Information Center. AASV News Archive. SVV oral fluids testing sheds light on distribution. Published September 16, 2015. Available at https://www.aasv.org/news/story.php?id=8361. Accessed 18 July 2016.
* Non-refereed references.