| Original research | Peer reviewed |
Cite as: Stricker AM, Polson DD, Murtaugh MP, et al. Variation in porcine reproductive and respiratory syndrome virus open reading frame 5 diagnostic sequencing . J Swine Health Prod. 2015;23(1):18–27.
Also available as a PDF.
SummaryObjective: To assess porcine reproductive and respiratory syndrome virus (PRRSV) open reading frame 5 (ORF5) sequencing variation, within and among state diagnostic laboratories, that may contribute to observed differences in sequence homology among isolates. Materials and methods: PRRS virus-positive blood samples were collected from individual pigs on three different farms and submitted on three independent occasions to three diagnostic laboratories for PRRSV ORF5 nucleotide sequencing. The PRRSV isolates on each farm were genetically disparate. Vaccine viruses (Ingelvac PRRS MLV and Ingelvac PRRS ATP; Boehringer Ingelheim Vetmedica, Inc, St Joseph, Missouri) were submitted as positive controls. Results: Full-length ORF5 sequences were obtained from all samples. Positive-control vaccine virus sequencing was precise and highly accurate, with all laboratories on all occasions obtaining nearly identical sequences. The analytical specificity of field PRRSV sequencing was robust, with a median variation among laboratories for the same farm sample, across all pigs and submission dates, of one base difference per 603-base sequence (0.2%). Seventy-five percent of sequences had fewer than six base differences, and the greatest difference was 2.2%. However, 16% of samples in one submission from one farm appeared to be misidentified in the reports of one laboratory. Implications: Inter- and intra-laboratory ORF5 sequencing results are reproducible, reliable, and do not contribute significantly to estimated PRRSV diversity. Tracking errors may occur which can lead to confusion or inappropriate reaction by key decision makers. Submitters should retain aliquots of all samples to enable further investigation of a diagnostic error not related to the sequencing procedure. | ResumenObjectif: Évaluer les variations dans la séquence du cadre de lecture ouvert 5 (ORF5) du virus du syndrome reproducteur et respiratoire porcin (VSRRP), à l’interne et parmi les laboratoires de diagnostic d’état, qui pourraient contribuer aux différences observées dans les séquences d’homologie parmi les isolats. Matériels et méthodes: Des échantillons sanguins positifs pour VSRRP furent prélevés de porcs individuels sur trois fermes différentes et soumis à trois occasions indépendantes à trois laboratoires de diagnostic pour séquençage de l’ORF5 du VSRRP. Les isolats de VSRRP de chaque ferme étaient génétiquement disparates. Les virus vaccinaux (Ingelvac PRRS MLV et Ingelvac PRRS ATP; Boehringer Ingelheim Vetmedica, Inc, St Joseph, Missouri) furent soumis comme témoins positifs. Résultats: Les séquences complètes de l’ORF5 furent obtenues de tous les échantillons. Le séquençage des virus vaccinaux était reproductible et très précis, des résultats presqu’identiques étant obtenus par tous les laboratoires à toutes les occasions. La spécificité analytique du séquençage des échantillons de VSRRP du terrain était robuste, avec une variation médiane parmi les laboratoires pour l’échantillon de la même ferme, pour tous les animaux et dates de soumission, d’une différence d’une base nucléotidique par séquence de 603 bases (0,2%). Soixante-quinze pourcent des séquences avaient moins de six bases de différence, et la plus grande différence était de 2,2%. Toutefois, 16% des échantillons dans une soumission en provenance d’une ferme ont semblé être mal identifiés dans les rapports d’un des laboratoires. Implications: Les résultats de séquençage inter- et intra-laboratoire de l’ORF5 sont reproductibles, fiables, et ne contribuent pas significativement à la diversité estimée du VSRRP. Des erreurs de suivi pourraient se produire ce qui entrainerait de la confusion ou des réactions inappropriées par des décideurs clés. Les personnes soumettant des échantillons devraient conserver des aliquotes de tous les échantillons afin de permettre des études ultérieures en cas d’erreur diagnostique non reliée à la procédure de séquençage. | ResuméObjetivo: Valorar la variación de la secuencia del marco 5 (ORF5 por sus siglas en inglés) de lectura abierta del virus del síndrome reproductivo y respiratorio porcino (vPRRS), dentro y entre los laboratorios de diagnóstico estatales, que puedan contribuir a las diferencias observadas en la homología de secuencias entre aislamientos. Materiales y métodos: Se recolectaron muestras de sangre positivas al vPRRS de cerdos individuales en tres granjas diferentes y se enviaron en tres ocasiones independientes a tres laboratorios de diagnóstico para la secuencia de nucleótidos del ORF5 del vPRRS. Los aislamientos del vPRRS en cada granja eran genéticamente diferentes. Los virus de vacuna (Ingelvac PRRS MLV y Ingelvac PRRS ATP; Boehringer Ingelheim Vetmedica, Inc, St Joseph, Missouri) se enviaron como controles positivos. Resultados: Se obtuvieron secuencias de ORF5 completo de todas las muestras. La secuenciación del control positivo del virus de la vacuna fue precisa y muy exacta, todos los laboratorios en todas las ocasiones, obtuvieron secuencias casi idénticas. La especificidad analítica de la secuenciación del vPRRS de campo fue robusta, con una variación media entre laboratorios para la misma muestra de granja, entre todos los cerdos y fechas de entrega, de una base de diferencia por cada 603 bases (0.2%). Setenta y cinco por ciento de las secuencias tuvieron menos de seis bases de diferencia, y la mayor diferencia fue 2.2%. Sin embargo, 16% de las muestras en una entrega de una granja, parecen haber sido mal identificadas en los reportes de un laboratorio. Implicaciones: Los resultados de secuenciación ORF5 inter y entre laboratorio son reproducibles, confiables, y no contribuyen significativamente a la diversidad estimada del vPRRS. Pueden ocurrir errores de seguimiento que confundan o lleven a una reacción inadecuada de los responsables claves de toma de decisiones. Quienes envían muestras deberían retener alícuotas de todas las muestras para permitir investigaciones posteriores de un error de diagnóstico no relacionado con el proceso de secuenciación. |
Keywords: swine, porcine reproductive and respiratory syndrome, sequence, dendrogram, variation
Search the AASV web site
for pages with similar keywords.
Received: December 27, 2013
Accepted: July 14, 2014
One of the first questions asked at the onset of a clinical porcine reproductive and respiratory syndrome (PRRS) outbreak in a swine breeding herd is if the virus responsible is a new introduction or if it re-emerged from a previous resident field virus. An informed answer will help in determining if the farm experienced a new external virus introduction, indicative of a biosecurity breach, or if the persistent circulation of resident virus is responsible for an observed clinical episode.
PRRS virus (PRRSV) open reading frame 5 (ORF5) sequencing is commonly utilized as a means to help evaluate the origin, transmission, and circulation behavior of PRRSV within and among pig populations and regions. Using nucleotide and amino acid sequence data, percent ORF5 homology can be determined and a dendrogram generated to help determine relatedness of one virus to another in diagnostic samples. Results of field and experimental studies suggest, when two sequences are compared, a difference in identity greater than 2% to 3% is an indication they may not be closely related, although there is no general consensus on the amount of variation.1-3 With swine veterinarians often placing considerable importance on PRRSV sequence comparisons when investigating potential sources of virus exposure and developing a plan of action for farms with active PRRS infections, it is important to apply appropriate heuristic methods for sequence comparison. Given the real but poorly understood potential for sequencing process-related error, it cannot be assumed that the entire difference in sequence homology observed is attributable to actual differences in the viruses. Previous research has suggested that nucleic acid sequencing may be prone to various types and magnitudes of sequencing error, with the aggregate of errors contributing false diversity to the difference in homology between PRRSV isolates.3-6 PRRS virus is an RNA virus that is prone to undergo changes via mutation or recombination or both in infected pigs and populations.1,5,7,8 In one study, random technical errors accounted for up to half of the ORF5 sequence variation in individual PRRSV clones from the same pig.5
ORF5 is the most variable and immunologically relevant of the ORFs comprising the PRRSV genome, making it the preferred region to sequence to assess PRRSV genetic variability.5,7,9 However, unless swine veterinarians develop an appreciation for the degree of sequencing process variation, they are at risk of interpreting a virus isolate as a new introduction when it is not, and implementing actions that they would otherwise not recommend. The objective of this study was to address the hypothesis that PRRSV ORF5 sequencing variation within and among state diagnostic laboratories may contribute to differences in sequence homology among PRRSV isolates.
Materials and methods
This study did not require ethical review because the activities comprised part of a periodic, routine diagnostic monitoring program and did not involve animal experimentation.
Blood was collected via venipuncture in 9-mL serum separator tubes from six or seven suspected PRRS-positive pigs at three geographically separate wean-to-finish farm locations as part of routine veterinary care and disease surveillance. Six tubes were collected from each pig sampled. The tubes were placed on ice and transported to a sample-processing facility (Suidae Health and Production, Algona, Iowa). Tubes then were centrifuged at 398g for 10 minutes. Recovered serum was pooled for each pig. From this pool, approximately 1-mL aliquots were placed into labeled snap-cap tubes that were placed in a freezer and held at -80°C. One aliquot from each pig was sent to Iowa State University Veterinary Diagnostic Laboratory (Ames, Iowa) for confirmation of PRRS-positive status via a PRRSV reverse-transcriptase polymerase chain reaction (RT-PCR). On the basis of these results, the three PCR-positive pigs with the lowest threshold cycle (Ct) values (signifying the highest virus concentrations) were chosen from each farm to be included in the remaining phases of the study.
Positive controls with known sequences were also created using two commercially available modified-live PRRSV vaccines (Ingelvac PRRS MLV and Ingelvac PRRS ATP; Boehringer Ingelheim Vetmedica, Inc, St Joseph, Missouri). To maximize the likelihood of identical control sequences, control viruses were obtained from a single 50-dose bottle for each vaccine virus. Vaccine was mixed with serum collected from pigs on a farm with a PRRS-negative testing history. The PRRS-negative status of this serum was confirmed prior to mixing with the control viruses via PRRSV RT-PCR testing at the Iowa State University Veterinary Diagnostic Laboratory.
On submission day 0, a total of 33 serum samples consisting of three tubes from each of three pigs at each of three farms (FF, PE, and TNT), along with three tubes from each of the controls, were packaged on ice and shipped for overnight delivery to each of three state diagnostic laboratories with a history of handling a high volume of swine diagnostic samples, including PRRSV ORF5 sequencing. Each sample was assigned a number between 1 and 33 using a random numbers table generated in a commercial spreadsheet program (Excel 2007; Microsoft Corporation, Redmond, Washington). Coding was assigned according to the diagnostic laboratory, submission, farm, and pig (ie, X1-FF-A1 indicated laboratory X, submission 1 of 3, Farm FF, pig A, tube 1 of 3). The entire submission process was repeated two more times at approximately 30 and 60 days after the first submission. The total number of field and vaccine virus samples submitted was 297.
Samples were submitted as “known PRRS-positive” to each laboratory and a request was made for all samples to be submitted directly for ORF5 sequencing. This was done as a cost-savings step to eliminate the need for a screening PCR and to accommodate direct testing on the ORF5 PCR for sequencing. Raw sequencing data was requested from each laboratory for analysis. All of the raw ORF5 nucleotide sequencing data was collected from each of the diagnostic laboratories and aligned using the Clustal W “slow-accurate” method included in the commercially available software Lasergene DNAStar Megalign version 8.1.2 (Madison, Wisconsin). A master dendrogram and homology table containing all resulting field virus and vaccine references was generated using the Lasergene software. For the purposes of the similarity analysis, paired sequence comparisons that were expected to be identical, since they were from the same sample, were defined as two sequences having greater than or equal to three of 603 nucleotide differences, equivalent to a homology of 99.50% or greater, since sequencing process-related errors of up to 0.50% were assumed possible but considered non-significant to the barn-level decision-making process and intervention-plan process by the veterinarian and producer. Further, paired sequence comparisons that were expected to be identical but that had less than 97.00% homology were defined as “outliers.”3 The results were attributed to laboratory processing errors rather than sequencing errors.
Upon completing alignment of the raw data, a visit was made to each of the three participating diagnostic laboratories to discuss the results, as well as to gain further insight into the sequencing process from receiving a sample to reporting results.
Data was analyzed using descriptive statistics. Specific comparisons were made using a multiple comparison of proportions test, specifically Tukey’s honest significant difference (HSD) test, in MULTPROP.mac (Minitab 17.1.0, Minitab Inc, State College, Pennsylvania).
Results
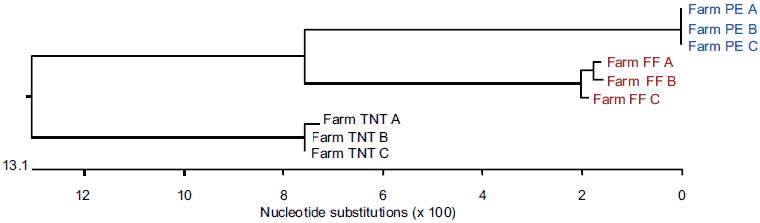
Genetically distinct viruses differing by more than 13% in nucleotide sequence identity were present on each of the three farms, as shown in Figure 1 and confirmed by direct pairwise comparison (data not shown). Among the entire set of 297 sequences in this study, all of the sequences on one farm had 100% nucleotide agreement, whereas the sequences from the other two farms differed in the range of 0.2% to 0.8%, ie, from one to five bases per 603 bases in ORF5.
Figure 1: Dendrogram from blood samples positive for porcine reproductive and respiratory syndrome (PRRS) virus collected from individual pigs (A, B, and C) on three different farms (FF, PE, and TNT) and submitted on three independent occasions to three diagnostic laboratories (X, Y, and Z) for PRRS virus open reading frame 5 nucleotide sequencing.
Vaccine control comparisons
Analysis of two independent positive-control vaccine strains was used to estimate intra- and inter-laboratory diagnostic sequencing variation. As shown in Table 1, the vaccine controls had 100% nucleotide agreement regardless of submission time for Laboratory X and Laboratory Y. Likewise, there was 100% nucleotide sequence agreement between laboratories X and Y on all pair-wise comparisons. Laboratory Z had 100% nucleotide agreement on 58 of 72 positive-control pair-wise comparisons (80.6%), and 14 sequences differed by one nucleotide from the consensus (Table 1). Interestingly, all Laboratory Z MLV vaccine control sequences differed at one position, base 8, from all sequences obtained in Laboratory X and Laboratory Y, and ATP vaccine sequences from Laboratory Z differed from all sequences reported from Laboratory X and Laboratory Y at positions 11 and 599. Thus, 100% agreement was obtained in all cases between Laboratory X and Laboratory Y, but neither showed perfect agreement with any vaccine sequence reported from Laboratory Z.
Table 1: PRRS vaccine control sequence agreement within and among laboratories X, Y, and Z across submissions*
| Comparison | n | Percent with 100% identity | Average % identity | Minimum % identity | |
| Laboratory 1 | Laboratory 2 | ||||
| X | X | 72 | 100.00a | 100.00 | 100.00 |
| Y | Y | 72 | 100.00a | 100.00 | 100.00 |
| Z | Z | 72 | 80.60b | 99.96 | 99.80 |
| X | Y | 162 | 100.00a | 100.00 | 100.00 |
| X | Z | 162 | 0.00b | 99.74 | 99.70 |
| Y | Z | 162 | 0.00b | 99.74 | 99.70 |
* Study described in Figure 1. Analysis of two independent positive-control vaccine strains was used to estimate intra- and inter-laboratory diagnostic sequencing variation. Positive controls with known sequences were created using two commercially available modified-live PRRS virus vaccines (Ingelvac PRRS MLV and Ingelvac PRRS ATP; Boehringer Ingelheim Vetmedica, Inc, St Joseph, Missouri). Two three-way comparisons were made: within laboratory (XX, YY, ZZ) and between laboratories (XY, XZ, YZ).
a,b Values within a column with differing superscripts are significantly different (P < .05; Tukey’s HSD test).
PRRS = porcine reproductive and respiratory syndrome; HSD = honest significant difference.
Wild-type comparisons
The total variation in diagnostic sequencing results in the first set of submitted samples (referred to as day 0) is shown in Figure 2. The samples from all three farms submitted to both Laboratory X and Laboratory Z clustered with the farm viral sequence as expected, as did samples from two of three farms submitted to Laboratory Y. The results were expected because the samples had been sequenced previously and were known to cluster within each farm as shown in Figure 1. However, at Laboratory Y, Farm FF samples showed discrepancies, with only one of nine samples clustering as expected. Seven of the eight Laboratory Y discrepant samples were grouped with the TNT cluster and one was grouped with the PE cluster. In the second submission set, all sequences from Laboratory X and Laboratory Z clustered as expected, as did two of the three sequence sets for Laboratory Y (Figure 3). However, at Laboratory Y, Farm FF samples again showed discrepancies, with only one of the nine samples clustering as expected. Seven of the eight Laboratory Y discrepant samples were grouped within the TNT cluster (Figure 3). The same pattern was observed with the third submission as well: seven of nine Farm FF samples submitted to Laboratory Y clustered as expected, and two samples were grouped with the TNT cluster (data not shown). The original dendrogram demonstrated greater than 15% difference in homology between Farm TNT isolates and Farm FF isolates, and greater than 13% difference in homology among isolates from Farm PE (Figure 1).
Figure 2: Dendrogram of open reading frame 5 sequences obtained from the first laboratory submission event (submission events described in Figure 1). Yellow highlighting represents sample group discrepancies.
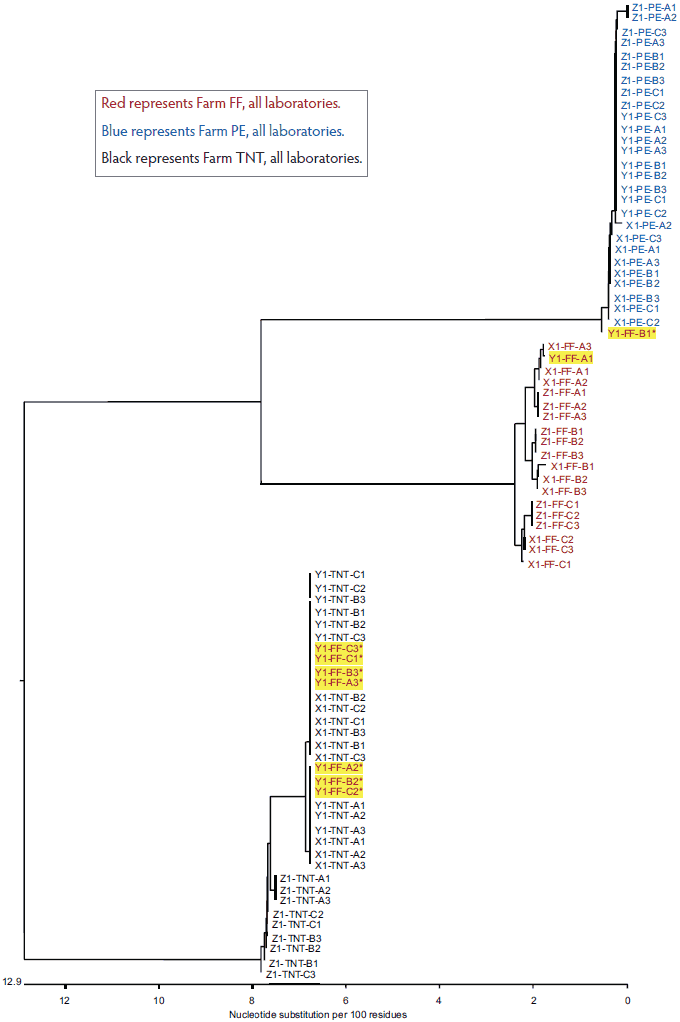
Red represents Farm FF, all laboratories.
Blue represents Farm PE, all laboratories.
Black represents Farm TNT, all laboratories.
Figure 3: Dendrogram of open reading frame 5 sequences obtained from the second laboratory submission event (submission events described in Figure 1). Yellow highlighting represents sample group discrepancies.
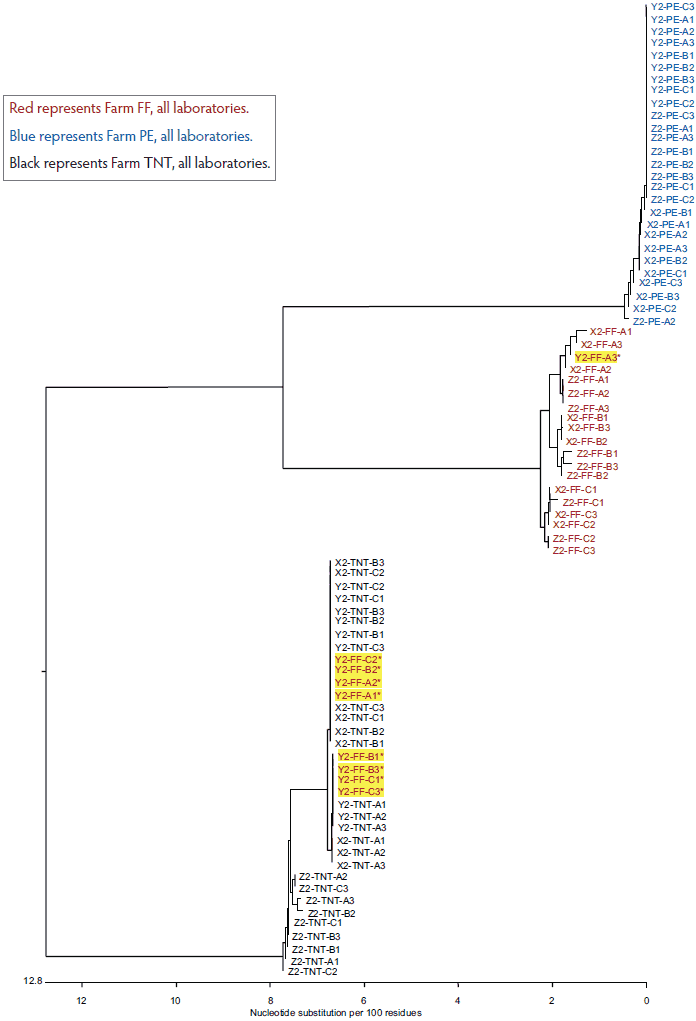
Red represents Farm FF, all laboratories.
Blue represents Farm PE, all laboratories.
Black represents Farm TNT, all laboratories.
After presenting the data and scope of the research to Laboratory Y, a request was made by the laboratory to analyze a fourth submission. Surprisingly, as shown in Figure 4, two sequences appeared to be identical to the Farm TNT isolates even though one was identified as Farm PE and the other was identified as Farm FF.
Figure 4: Dendrogram of open reading frame 5 sequences obtained from the fourth submission to Laboratory Y, performed because of discrepancies (yellow highlighting) in the results of the first three submissions (submissions described in Figure 1).
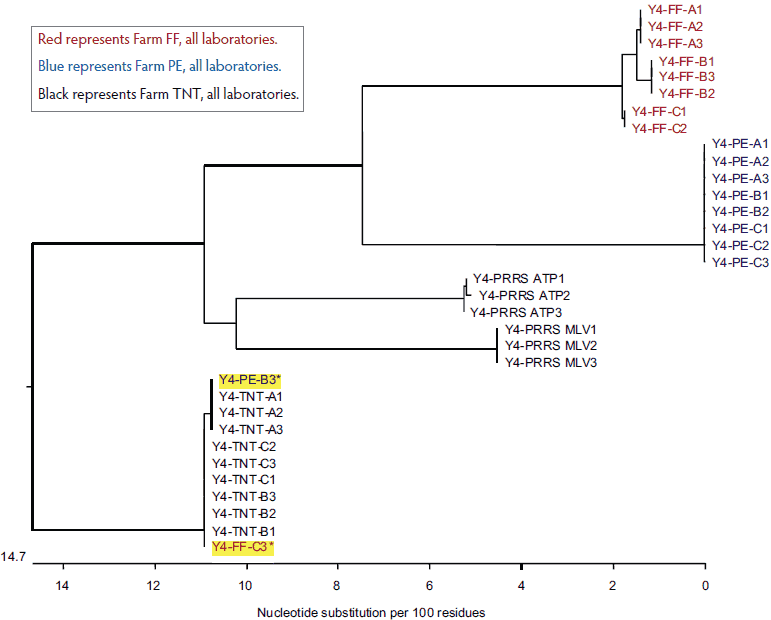
Red represents Farm FF, all laboratories.
Blue represents Farm PE, all laboratories.
Black represents Farm TNT, all laboratories.
It appeared that submission or reporting errors occurred in Laboratory Y, since clustering results from Laboratory Y sequences were indistinguishable from those obtained from Laboratory X and Laboratory Z (Figures 2 and 3). Therefore, to address the specific issue of sequencing variability and reliability, field-sample sequences were analyzed by phylogenetic cluster. When compared across submissions with all other variables controlled, Laboratory X and Laboratory Z met the sequencing fidelity criteria of greater than or equal to 99.5% homology (fewer than or equal to three base differences from the consensus sequence) across all submitted samples. In comparison, Laboratory Y met the same reliability criteria for sequencing field isolates 84.3% of the time (Table 2).
Table 2: Reliability comparison for all PRRS field-virus submissions*
| Laboratory | Meets 99.5% identity criterion† | n (%) | Average % identity | Minimum % identity |
|---|---|---|---|---|
| X | 0 | 0 (0) | NA | NA |
| 1 | 324 (100)a | 99.98 | 99.70 | |
| Y | 0 | 51 (15.7) | 83.07 | 81.50 |
| 1 | 273 (84.3)b | 99.98 | 99.50 | |
| Z | 0 | 0 (0) | NA | NA |
| 1 | 324 (100)a | 99.93 | 99.70 |
* Study described in Figure 1 and Table 1.
† Sequencing fidelity criteria: ≥ 99.5% homology (≤ 3 base differences from the consensus sequence) across all submitted samples; 0 = did not meet criterion; 1 = met criterion.
a,b Values with differing superscripts are significantly different (P < .05; Tukey’s HSD test).
PRRS = porcine reproductive and respiratory syndrome; HSD = honest significant difference.
The overall reproducibility of sequencing within laboratories was high, as shown in Figure 5. One hundred percent of field-virus sequences were greater than 99.5% identical within individual laboratories, ie, they had three or fewer nucleotide differences from the consensus sequence. Comparison of Laboratory X to Laboratory Y showed that their inter-laboratory variation was negligible (Figure 5). However, comparison of Laboratory Z to either Laboratory X or Laboratory Y showed lesser agreement of 33.3% (66.7% for ZX compared to 100% for YX) and 42.9% (57.1% for ZY compared to 100% for YX), respectively, for three or fewer base differences. The result suggested the presence of a consistent three- to five-base difference in sequencing results that was unique to Laboratory Z.
Figure 5: Distribution of non-consensus open reading frame 5 sequence variants in all farms within laboratories (excludes outliers and vaccine controls) in submissions described in Figure 1.
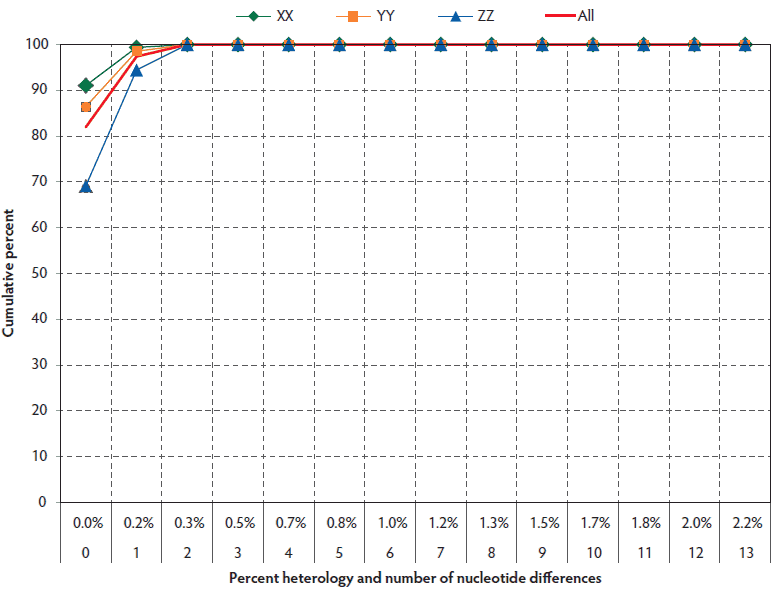
Analysis of variation across submissions in the same laboratory, farm, and pig, excluding vaccine controls and outliers, showed that all laboratories met the 99.5% homology criteria in 100% of pair-wise comparisons (Table 3).
Table 3: Wild-type PRRS open reading frame 5 sequencing variation within laboratories but among submissions*
| Comparison | n† | Percent with ≥ 99.5% identity | Average % identity | Minimum % identity | |
|---|---|---|---|---|---|
| Laboratory 1 | Laboratory 2 | ||||
| X | X | 324 | 100.00 | 99.98 | 99.70 |
| Y | Y | 324 | 84.26 | 97.31 | 81.50 |
| 273 | 100.00 | 99.98 | 99.70 | ||
| Z | Z | 324 | 100.00 | 99.93 | 99.70 |
* Study described in Figure 1 and Table 1.
† Excluding vaccine controls (described in Table 1); for Laboratory Y, results are presented both with outliers included and with 51 outliers excluded.
Alternative alignment methods occasionally used in the DNASTAR Megalign analysis can result in different results even when the same data are analyzed. Three common multiple alignment methods, Clustal W, Clustal V, and Jotun-Hein, are used to assemble and compare ORF5 sequences. To determine if differences in alignment method contributed to sequencing variation, the three methods were compared using the full dataset. The average nucleotide discrepancy was far less than one nucleotide across comparisons with all three alignment methods. The maximum percent discrepancy was 0.3% when comparing Clustal V and W, with 1.9% having at least one discrepant nucleotide. Comparison of Clustal W and Clustal V to Jotun-Hein revealed a maximum discrepancy of 0.7%, with at least one discrepant nucleotide in 13.5% and 14.8% wild-type sequences, respectively. Thus, the relative effect of differences in alignment method was insignificant.
Discussion
Variation in percent identity between PRRSV samples can be explained, in part, by differences in the wild-type viruses, even within a single pig sample.5 However, some deviations may also be explained by variation in the sequencing process or process execution or both within and among the laboratories themselves that may include both biological and technical factors. Since the study was focused on the potential contribution of technical variation that might result in misinterpretation of data, several steps were taken to minimize or remove within-pig variation.
All of the serum representing an individual pig in this study was taken from the same pig at the same time on the same day to account for the potential to have multiple PRRSV variants, or quasispecies, coexisting within individual pigs.5 The presence of sequence variation in the vaccine controls, which were not amplified in pigs, further indicates that biological variation was not the source of sequence differences. Hence, it is likely that the sequencing process itself contributed variation to the final result.
Lasergene DNAStar Megalign version 8.1.2 software was utilized by all three of the laboratories represented in this study. The software generates a table of sequence distances with percent identity on the x-axis and percent divergence on the y-axis. Percent identity compares the sequences directly, without taking phylogeny into account. Percent divergence differs in that it is not simply the inverse of the percent identity. Rather, the program uses an algorithm to calculate percent divergence that takes into account the sequence pairs in relation to the reconstructed phylogeny. Therefore, subtracting percent homology from 100 to calculate percent divergence is inappropriate, as is subtracting percent divergence from 100 to calculate percent homology. More importantly for this analysis, percent homology is consistent with further nucleotide-by-nucleotide analysis among sequences, whereas percent divergence is not. Comparison of sequences using identity and divergence interchangeably was avoided to eliminate it as a possible source of variation.
During the sequencing process, each nucleotide is identified by its own dye, which fluoresces at a specific peak wavelength. The result is a trace file graph called an electropherogram, which contains colored peaks, each representing one nucleotide in the sequence. To improve the accuracy of the sequence, multiple reads (two or three depending on the laboratory) were conducted and compared to yield a consensus sequence. To achieve the consensus sequence, the software aligned the various reads and used an algorithm to assign a base identity to each position. If the reads showed conflicting bases, the computer assigned a letter specific for combinations of any two or three possibilities, depending on which bases gave a peak at the same position. For example, if the conflict was between an A and a T, the letter W was assigned. If the peak was completely ambiguous, an N or X was assigned to the position and it was referred to as a “no-call”. Some diagnostic laboratories assign a technician to manually proof-read the consensus sequence, since machine reading errors occasionally occur. Variation among laboratories in manual proofreading contributes to inter-laboratory variation in results and may account for a portion of the differences observed here.
When Megalign aligns two sequences, it compares the base at each of the 603 positions that make up the North American PRRSV ORF5. Each position where a difference occurred was noted and used to generate a pairwise identity matrix for each alignment. Although the use of degenerate coding preserves more information, it is a source of variation between laboratories that will result in inter-laboratory differences in sequence analysis. For example, if two sequences both had discrepancies at the same positions, but one laboratory designated them all as “N” while the other designated them according to the universal degenerate code classification, the program would indicate a difference in homology between the two sequences at that position. One disagreement automatically results in a 0.2% difference in identity between two likely identical strains.
Sequences that contain multiple ambiguities should not be relied on for diagnostic interpretation, since they are indicative of a poor-quality sample, insufficient sample, or a true mixture of viruses that can be obtained from pooled sera. Most laboratories will report a failure to achieve more than one read or a sequence that contains several no-calls.
Another way to identify artificial variation in sequence analysis is to request the raw data text file or, preferably, the electropherogram. Typically, this will include the 603 base pairs that make up North American PRRSV strains or the 606 base pairs that make up European PRRSV strains. Insertions or deletions occasionally occur that vary the number of base pairs by a multiple of three, which increases or decreases the number of amino acids by a multiple of one, since three bases encode one amino acid. This biological variation contrasts with technical variation that may occur if untrimmed sequences, which vary in length due to extra bases outside of ORF5, are included in the analysis. This may be reflected as a difference in homology when none, in fact, exists.
The pronounced inter-laboratory disagreements were associated with three bases that were consistently the same within each of the three laboratories, yet different in Laboratory Z compared to laboratories X and Y. Since the differences were unambiguous, systematic, and not random, it indicates the presence of a highly reproducible difference in the sequencing process of one laboratory, such as in primers or kit chemistries or sequencing technology.
The specific cause(s) for the discrepancy in results between identical samples for Laboratory Y could not be determined. Without these discrepancies, Laboratory Y results would have been comparable to those of laboratories X and Z. Possible reasons for the notable outliers include sample cross-contamination during processing prior to submission to the laboratory or sample cross-contamination during the diagnostic-laboratory testing process. Errors in sample processing prior to submission were eliminated, since samples were drawn from the same tubes for all laboratories, yet the errors were confined to only one laboratory. It is reasonable to conclude that errors were introduced in sample handling or recording of data. Further research into sample handling prior to and after submission to the diagnostic laboratory would be expected to identify the source of error and enable its correction. The key finding here is that the sequencing method itself is reliable and is not a source of variation that could lead to misinterpretation of data and decision making.
Implications
• Under the conditions of this study, PRRSV ORF5 sequencing technology is robust and does not contribute significantly to genetic variation in phylogenetic analysis.
• Sample handling, processing, and other unidentified factors among laboratories may contribute substantially to observed sequence variation and, in turn, estimated PRRSV diversity.
• Veterinarians must be aware of the factors that can lead to process-related differences in sequence results.
• Occasional diagnostic errors can occur which may lead to confusion or inappropriate reaction by key decision makers. Submitters should retain aliquots of all samples to enable further investigation of unexpected variation.
Acknowledgements
The research was supported by an Advanced PRRS Research Award from Boehringer Ingelheim Vetmedica to Dr Stricker.
Conflict of interest
Dr Dale Polson was employed by Boehringer Ingelheim Vetmedica, Inc, at the time of the study.
Disclaimer
Scientific manuscripts published in the Journal of Swine Health and Production are peer reviewed. However, information on medications, feed, and management techniques may be specific to the research or commercial situation presented in the manuscript. It is the responsibility of the reader to use information responsibly and in accordance with the rules and regulations governing research or the practice of veterinary medicine in their country or region.
References
1. Yoon KJ, Chang CC, Zimmerman JJ, Harmon KM. Genetic and antigenic stability of PRRS virus in persistently infected pigs: clinical and experimental prospective. Adv Exp Med Biol. 2001;494:25–30.
2. Yuan S, Mickelson D, Murtaugh MP, Faaberg KS. Complete genome comparison of porcine reproductive and respiratory syndrome virus parental and attenuated strains. Virus Res. 2001;79:189–200.
*3. Polson DD, Baker RB, Philips R, Hotze B. Distribution characteristics of PRRS virus ORF5 sequences in a large production system applying intensive vaccination. Proc IPVS Cong. Durban, South Africa. 2008;2:134.
4. Malet L, Belnard M, Agut H, Cahour A. From RNA to quasispecies: a DNA polymerase with proofreading activity is highly recommended for accurate assessment of viral diversity. J Virol Methods. 2003;109:161–170.
5. Goldberg TL, Lowe JF, Milburn SM, Firkins LD. Quasispecies variation of porcine reproductive and respiratory syndrome virus during natural infection. Virology. 2003;317:197–207.
6. Bracho MA, Moya A, Barrio E. Contribution of Taq polymerase-induced errors to the estimation of RNA virus diversity. J Gen Virol. 1998;79:2921–2928.
7. Chang CC, Yoon KJ, Zimmerman JJ, Harmon KM, Dixon PM, Dvorak CMT, Murtaugh MP. Evolution of porcine reproductive and respiratory syndrome virus during sequential passage in pigs. J Virology. 2002;76:4750–4763.
8. Rowland RR, Steffen M, Ackerman T, Benfield DA. The evolution of porcine reproductive and respiratory syndrome virus:quasispecies and emergence of a virus subpopulation during infection of pigs with VR-2332. Virology. 1999;259:262–266.
9. Shi M, Lam TTY, Hon CC, Hui RKH, Faaberg K, Wennblom T, Murtaugh M, Stadejek T, Leung FCC. Molecular epidemiology of PRRSV: A phylogenetic perspective. Virus Res. 2010;154:7–17.
* Non-refereed reference.